
Primary screening & high throughput automated patch clamp
Patch clamp or fluorescent assays for primary screening
Fluorescent and luminescent assays have been the default choice for primary screening for decades. However, the introduction of high-throughput automated patch clamp (APC) platforms and their ability to run unattended overnight runs of up to 16 plates make patch clamping an appealing alternative.
Drug discovery remains a trial-and-error exercise in the quest for the next drug. Once a target has been identified, the next step is often to screen compound libraries to generate leads for later drug candidates. This efficiency of the screen is influenced basically by two parameters:
- Number of experiments per time unit
- Degree of the usefulness of results from the screening
While 1) is easy to measure, 2) takes more insight to evaluate. In most discovery processes, the flow can be described with the funnel depicted in Figure 1. The high throughput has been followed by secondary or confirmatory screen, lead optimization, safety studies and profiling. When targets are ion channels, everything from secondary screening has been patch-clamp, but the high throughput screen has been synonymous with fluorescence-based technologies due to the low throughput. Fluorescence-based technologies have high performance on parameter 1
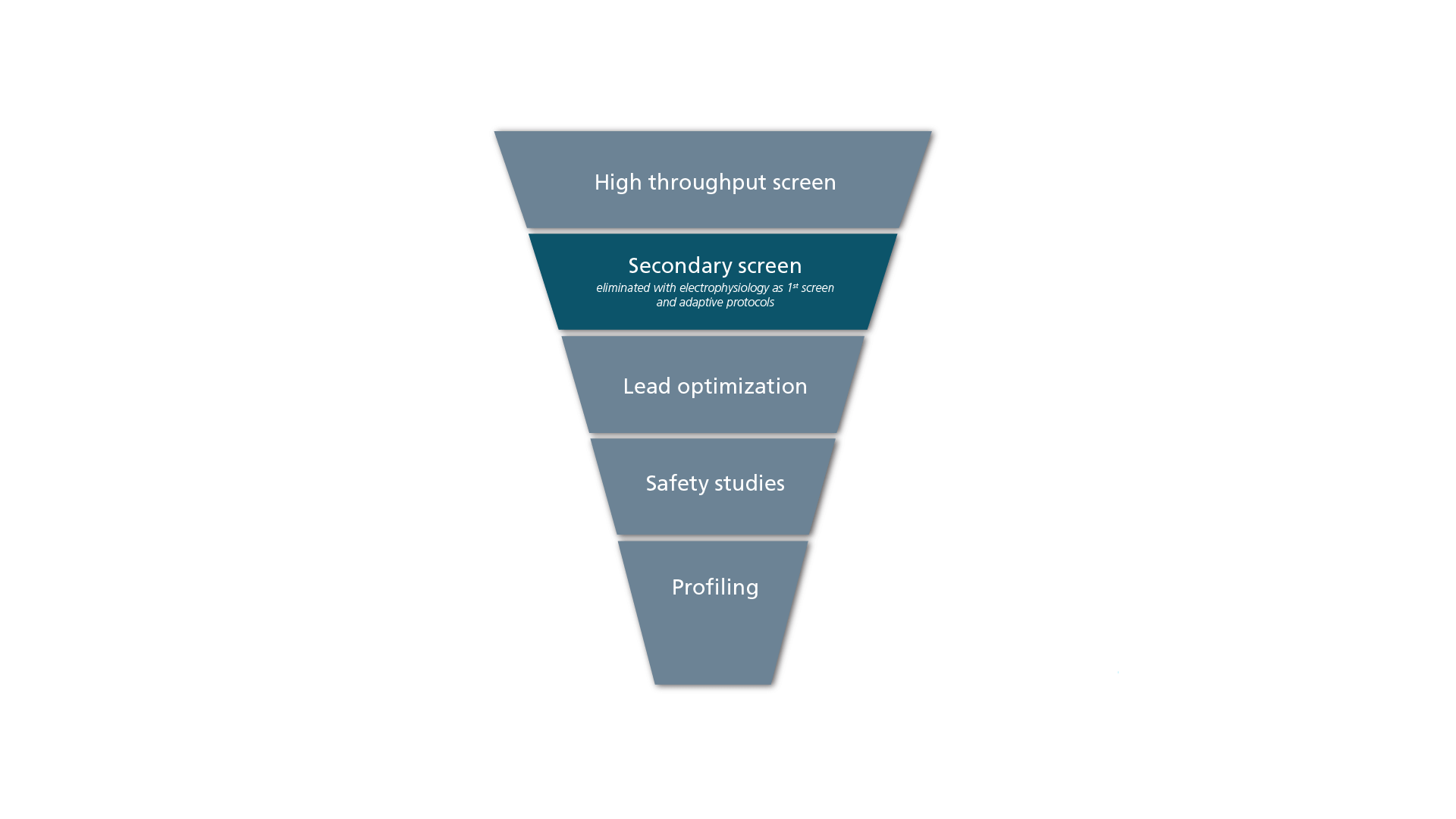
By performing the primary screen using automated patch clamp it is possible to omit the secondary screen, saving both money and time in your drug discovery process
With the introduction of high-throughput APC solutions, it has been possible to conduct electrophysiology as primary screening and therefore omit the secondary screen, simply because e-phys confirmation is already done with the primary screening.
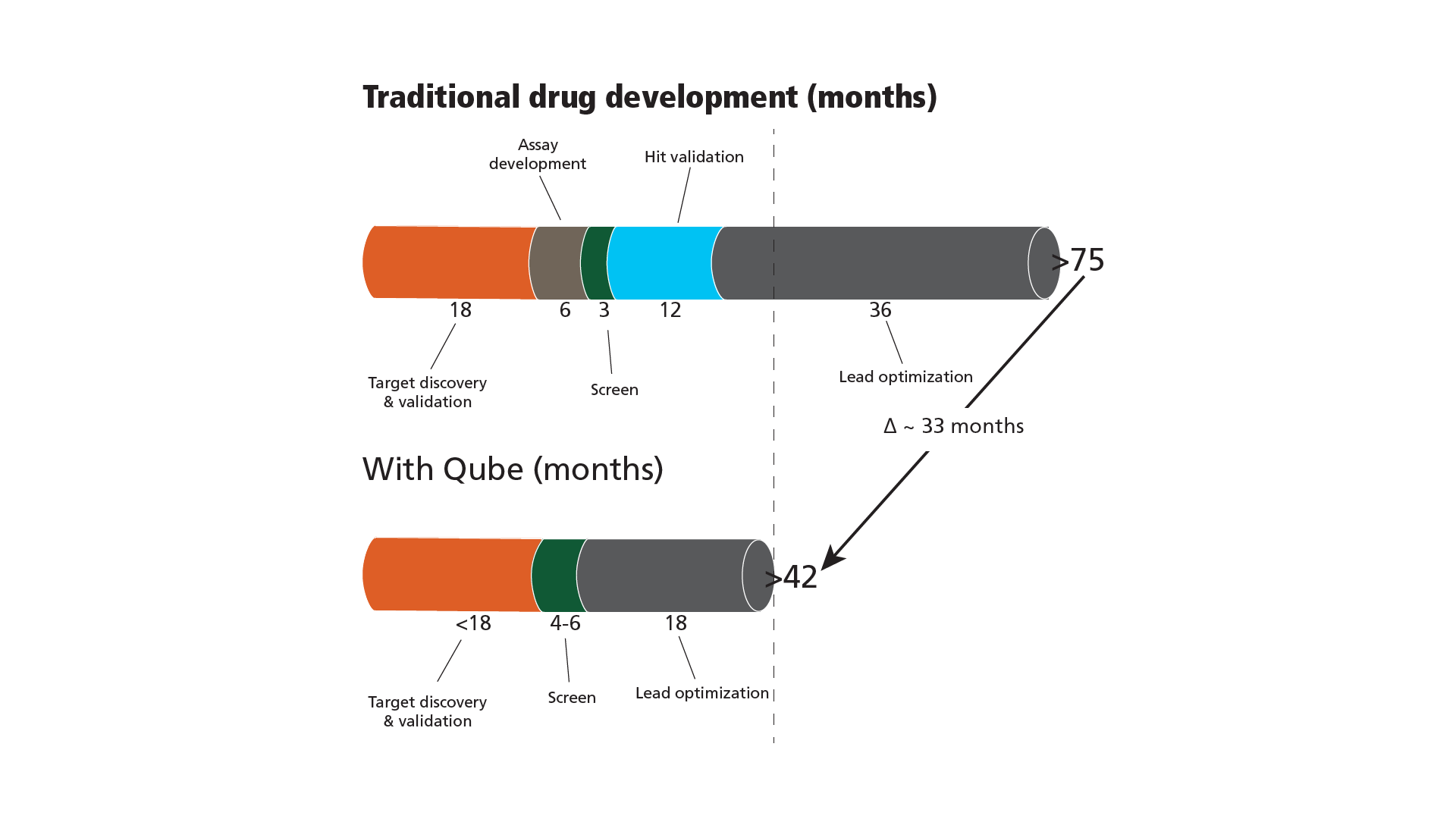
Using an APC screen, the price per data point is slightly higher since fluorescent assays. While fluorescent assay are around 5-10 cents per well, APC screening is around 20-30 cents/well, so the cost of an APC screen is admittedly higher. However, consider the entire in vitro part of the drug discovery process as depicted in Figure 2, where “Traditional….” means using fluorescence-based technology as primary screening.
The target discovery and validation are the same for the two processes. The assay development takes some time to find, e.g. Ca2+-dyes, membrane potential sensing dyes etc., for the FLIPR-assay. In contrast, the Qube APC is a patch-clamp instrument and does the same during the target discovery and validation. The screening part is faster on the FLIPR, but APC will more than catch up since:
- No need for hit validation (already patch clamp)
- Better hits/leads since the data are genuine ion channel recordings in contrast to indirect measures of downstream/ secondary messenger systems when using fluorescence-based technology.
In particular, parameter b) means that the medicinal chemists get better data to work with. They get the same type of data when the refined molecules are synthesized and tested since that is also done with patch-clamp. This means that both the more sophisticated on-target effects, such as mode-of-action and subtype selectivity and the off-target effects, like cardiac liability, are available very early in the drug discovery and will ensure a faster and more cost-efficient process. Looking at Figure 2 and imagining that the time saved in using APC as primary screening is converted into prolonged patent life once the molecule is developed into the final drug will tremendously enhance the value of using APC upfront.
Concentration response with much fewer wells
Do you run the usual 22-point concentration-response experiments on your FLIPR to determine IC50 values? That, of course, fits with the compound plate layout you likely have, but it also requires one assay plate with seeding of cells for every compound plate, and most importantly, you don’t have the control cell for every concentration. Consider running such an assay with a cumulative application on an automated electrophysiology platform:
- All 22 concentrations will have the same cell response as control
- All 22 concentrations will only use one well instead of 22
- The electrophysiological read-out is direct on the ion channel and hence more predictive
Reference:
Doesn't it take long to screen with electrophysiology?
Admittedly, automated electrophysiology is not as fast as fluorescence-based screening. Notably, e-phys so far only come in 384-format and not 1536-format, but automated e-phys can run unattended, so that makes up for some of it:
- ≥8 hours run without any need for personnel around the instrument
- >9,000 wells assayed unattended
- >25,000 wells assays per 24 hours
- Z’-values that support large screening windows
- The electrophysiological read-out is direct on the ion channel and hence more predictive.
Reference:
How do you best help your chemist – faster hit to lead?
After performing your initial screening on a fluorescence-based instrument, you often need to confirm this by electrophysiology. Why not do that, to begin with? Utilize your expertise in running an extensive screening campaign with all the logistics required for compound handling. Get the output values of pharmacological effects with high-fidelity electrophysiology, which can go directly to your chemists’ modelling program. In other words, faster from hit to lead.
- More precise pharmacology with adaptive protocols
- More relevant questions asked of your molecules
- More subtle effects of your molecules can be pursued
Reference:
How do you help your chemist the best way – better leads?
In the ideal world, modelling could generate the molecule you want to develop for any disease. In the real world, we still need data; the more predictive data, the better. When interrogating ion channels, the best data come from electrophysiology. And you can get that without jeopardizing the need for speed. With 384-format high-fidelity electrophysiology, you can run unattended; your chemist will always have the data needed in due time, and more importantly, with the data content for creating a better drug:
- State-dependent mode of action
- on/off rates
- IC50 values
- All of the above at any temperature between 10-40◦C
Reference:
Early de-risking of cardiac liability, hERG and NaV1.5
When do you take care of investigating the cardiac liability of your compounds? Wouldn’t have that early in the drug development and for multiple targets be great? You can get that in one operation with electrophysiology on, e.g. hERG and NaV1.5 and early on, find out if you need to direct your medicinal chemistry program around a problem.
- Take advantage of constant voltage clamp
- Take advantage of temporal resolution
- Take advantage of the multihole recording mode
- E.g. hERG and NaV1.5 pharmacology tested in the same assay
Reference:
Ligand-gated ion channels that desensitize – no problem
Some ligand-gated targets only allow short exposure to their ligand before desensitizing and don’t lend themselves to re-stimulation. That is a problem when your screening technology does not provide wash-out. But it is not a problem with electrophysiology with microfluidic flow channels and very short exposure times where the target can be stimulated multiple times and hence serve as their control read-out.
- Possible with repeated stimulation of fast-desensitizing ligand-gated channels like AchRα7
- Possible with cumulative concentration-response where each cell is its own control
- Possible to follow the kinetics of the target in control as well as the compound situation
- Possible to do all of the above in 384-format unattended
Reference:
- Characterization of the rapidly-desensitizing α7 nicotinic acetylcholine receptor using the Qube
Get in Touch
We strive to provide the best for our customers, and we are always ready to help. Please let us know if you have a question for us.